

Abstract
Background: PTLD is the most common malignancy, other than non-melanoma skin cancer, complicating solid organ transplantation (SOT) and has been one of the most commonly observed fatal consequences in SOT. The clinical presentations, management strategies, histologies, causes of death and outcomes are diverse (Dierickx D, Habermann TM. N Engl J Med 2018;378:549-62). The 2017 WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues has redefined the categories as non-destructive PTLDs (plasmacytic hyperplasia, infectious mononucleosis, and florid follicular hyperplasia), polymorphic PTLD, monomorphic PTLD: B-cell neoplasms (diffuse large B-cell lymphoma, Burkitt lymphoma, high-grade B-cell lymphoma, plasmablastic lymphoma, plasma cell myeloma, plasmacytoma, and other) and T-cell neoplasms (peripheral T-cell lymphoma NOS, hepatosplenic T-cell lymphoma, other), and classic Hodgkin lymphoma (CHL) PTLD. We report the outcomes and long-term follow-up of patients from a single institution based on these categories whose pathology was retrospectively reviewed and reclassified based on the WHO 2017 classification.
Methods: Patients with SOT who were diagnosed with PTLD at Mayo Clinic (Rochester, MN) were identified through the Mayo Clinic Lymphoma Data base and the University of Iowa/Mayo Clinic SPORE Molecular Epidemiology Resource (MER). The histology was re-reviewed in 80% of the cases (RLK) and classified according to the WHO Classification of Tumours of Haematopoietic an Lymphoid Tissues 2017. Cases were considered "PTLD, unclassified" if the histology could not be confidently classified based on pathology material available for review. Indolent small B-cell lymphomas were not included among the PTLDs except for EBV-positive marginal zone lymphoma. Cox proportional hazards models were used to assess the association of clinical factors in overall survival (OS).
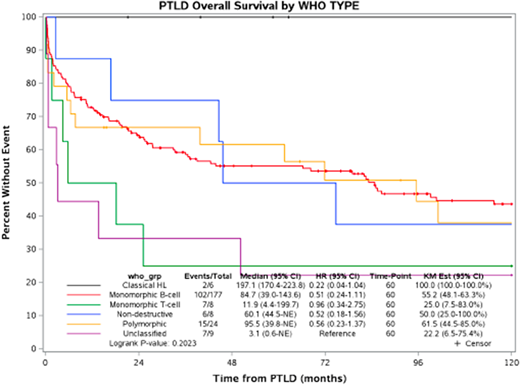
Results: 233 patients diagnosed with PTLD between 1987 and 2017 were identified. The median age at the time of diagnosis of PTLD was 54 years (range 16 to 84) with 85 patients (36%) over the age of 60. 156 (67%) were male. The transplanted organs were kidney (41%), kidney/pancreas (5%), liver (29%), heart (9%), lung (7%), and other (8%). PTLD occurred late (more than one year after transplantation) in 66%. There were 69 stage I, 19 stage II, 8 stage III, and 128 stage IV patients. 84% presented with extranodal disease. 21% had involvement of the engrafted organ. 64% of the patients developed a PTLD that was EBV positive by in situ hybridization. Initial approaches to management included reduction of immunosuppression (N=55), chemotherapy/immunochemotherapy (N=71), reduction in immunosuppression with rituximab (N=58), single agent rituximab (N=14), and radiation therapy (N=5). At a median follow-up of 87 months (range 9-289), 139 (60%) patients had died. All six CHL-PTLD patients are alive, two of whom had an event. The median overall survival (OS) was 85 months (95% CI: 39-144) in 177 monomorphic B-cell lymphoma PTLD, 95.5 months (95% CI: 40-not reached) in 24 polymorphic PTLD, and 60 months in 8 non-destructive PTLD cases. In contrast, the median overall survival was 12 months (95% CI: 4-200) in 8 monomorphic T cell and 3 months (95% CI: 1-unreached) in 9 unclassified PTLD cases.
Conclusion: PTLD is a heterogeneous group of immunodeficiency-associated lymphoproliferative disorders. The overall survival in non-destructive, polymorphic, and monomorphic PTLD were similar. Monomorphic T/NK cell types had inferior outcomes.
Maurer:Morphosys: Research Funding; Nanostring: Research Funding; Celgene: Research Funding. Ansell:Celldex: Research Funding; Merck & Co: Research Funding; Takeda: Research Funding; LAM Therapeutics: Research Funding; Seattle Genetics: Research Funding; Trillium: Research Funding; Bristol-Myers Squibb: Research Funding; Affimed: Research Funding; Regeneron: Research Funding; Pfizer: Research Funding. Cerhan:Celgene: Research Funding; Nanostring: Research Funding; Jannsen: Other: Scientific Advisory Board.

